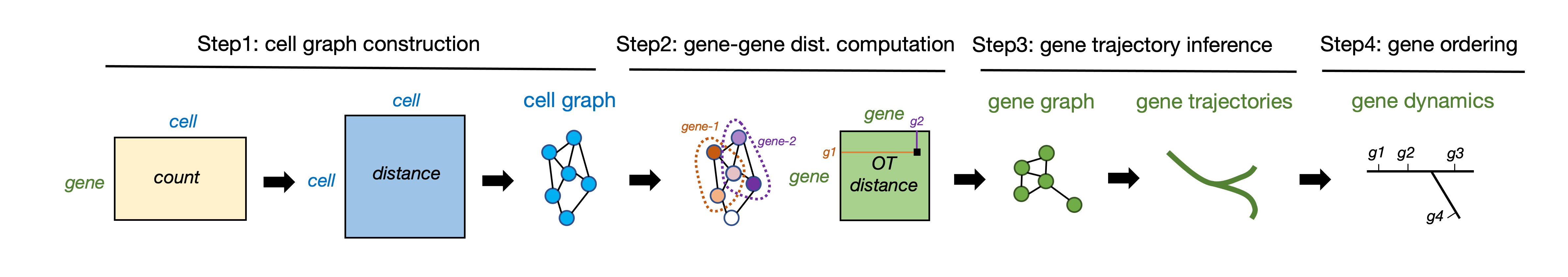
GeneTrajectory is a method for inferring gene trajectories in scRNA-seq data, which facilitates understanding of gene dynamics underlying biological processes. The major workflow of GeneTrajectory comprises the following four main steps: * Step 1. Build a cell-cell kNN graph in which each cell is connected to its k-nearest neighbors. Find the shortest path connecting each pair of cells in the graph and denote its length as the graph distance between cells. * Step 2. Compute pairwise graph-based Wasserstein distance between gene distributions, which quantifies the minimum cost of transporting the distribution of a given gene into the distribution of another gene in the cell graph. * Step 3. Generate a low-dimensional representation of genes (using Diffusion Map by default) based on the gene-gene Wasserstein distance matrix. Identify gene trajectories in a sequential manner. * Step 4. Determine the order of genes along each gene trajectory.
Install
GeneTrajectory can be installed as follows:
install.packages("devtools")
devtools::install_github("KlugerLab/GeneTrajectory")Example tutorial
Please check GeneTrajectory tutorial.
References
References of GeneTrajectory functions can be found here.
Data used in the tutorial can be downloaded from Figshare.
Install GeneTrajectory-python
The easiest way is to create a virtualenv for gene_trajectory using reticulate
if(!reticulate::virtualenv_exists('gene_trajectory')){
reticulate::virtualenv_create('gene_trajectory', packages=c('gene_trajectory'))
}
reticulate::use_virtualenv('gene_trajectory')or to add to an existing virtualenv using
reticulate::py_install("gene-trajectory")In general (especially in a conda environment) it can be installed with pip as
The development version can be installed as
system(sprintf('%s -m pip install git+https://github.com/Klugerlab/GeneTrajectory-python.git', reticulate::py_exe()))This works both on virtualenv and conda.